SOP_data
Standard Operating Procedures
Project maintained by stajichlab Hosted on GitHub Pages — Theme by mattgraham
You got your full genome sequencing results from Illumina, now what?
Usually Dr. Stajich has access and downloads this into the cluster under shared/projects/.
01-Let’s run a diagnostic or FastQC on your reads
In order to tell if your sequence results were conclusive run the program FastQC. Our HPCC/cluster has it saved as a module. So load it in with module load fastqc.
I always recommend running the help option for every new program you encounter this way you can see all the options available.
Type in fastqc -h to view the multiple options this program has. \n

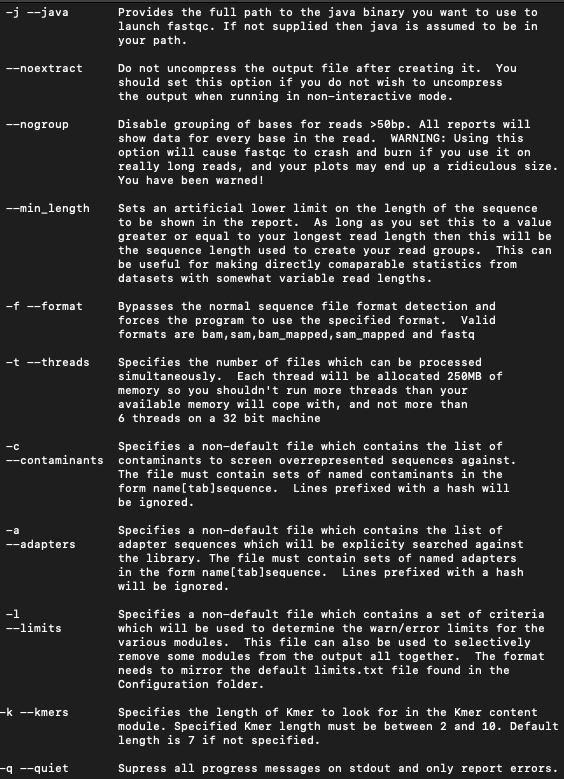
Make sure your files are in a proper folder. Create the folder FastQC in the same directory as your data.
When I run FastQC on my samples I run it through these options.
fastqc XX_1.fastq.gz XX_2.fastq.gz --noextract -o FastQC
All I’ve expressed here is - run fastqc on my forward run (1) and my reverse run (2), adding in –noextract allows my fastq.gz files to remain zipped.
We do this to conserve space, but also our assembly programs accept files in this format so there’s no need to unzip them.
My last step -o FastQC just directs my results into my FastQC folder, this way it is organized and away from the original data.
You can run this all on your directory or pass it through slurm. Though I’ve noticed the more you add in the script the more likely the code breaks.
As the code runs and finishes the files are created in FastQC. Direct yourself into the folder to observe the files.
Each read (forward and reverse) will have run separately and you will receive two files each (4 total). One being a zip file and the other an html file.
Find the suggestions on the HPCC webpage (http://hpcc.ucr.edu) on how to link your html files to your .html folder in your home page.
That way you can open up your cluster onto an html link and view the report.
001-How to read your diagnostic results
You got some nice figures on your html link- what does it all mean?
My main focus is the top summary figure.
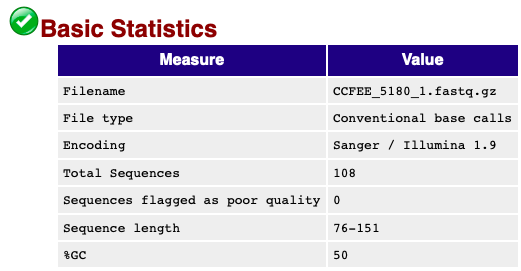
This is a bad quality read.
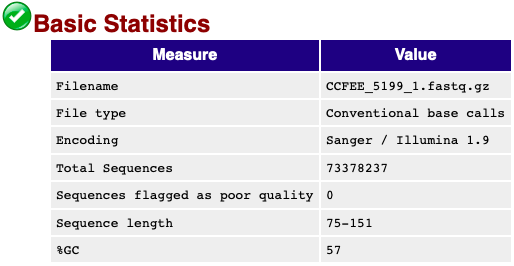
This one is much much better.
It gives you the total number of reads and that way you can see if the sequencing was successful or not. Look for checkmarks and see if your sequence lengths are good. If you are curious there is a FastQC Manual (https://rtsf.natsci.msu.edu/genomics/tech-notes/fastqc-tutorial-and-faq/) to help clarify your concerns.
02- Create your samples.csv
What’s this? It’ll be the document you direct your script to look into. It’ll have information you’ll be needing to help assemble your genomes. Basically you’ll be creating a basic text file that describes your strains.
In Dr. Stajich’s code he has a line that reads sed -n ${N}p $SAMPLEFILE | while read BASE STRAIN PHYLUM
What this line of code does is that it considers your N
(this will be the iteration your code is running in. If you’ve got more than one strain running it’ll be run as an array, described more later)
Whatever number your array is on, it will consider that line of code. I.E. N = 1, will read the first line, etc.
-BASE = I consider the Genus and species name of your organism. If you’re unsure of the species going for sp. is good too.
-STRAIN = Will be the short letter format of your reads. Usually it’ll be in a format you sent to the company who did your sequencing.
-PHYLUM = List the phylum of your strain!
Your document should look like this
Exophiala_xenobiotica,TK68,Ascomycete
Exophiala_xenobiotica,TK66,Ascomycete
..
Once complete it helps to do a quick wc -l samples.csv on your terminal to see how many lines this is. It will come in handy when you run them in an array.
It’s very important you take a look at Dr. Stajich’s scripts and follow along that sed line. It’ll help direct you on what your samples file should look like.
03- Orienting your workspace
Make sure you have your directory all set up to do your assembly analysis.
Have your Illumina sequence data in your input/ folder.
Have your samples.csv file sitting in the main directory where your input folder lives.
Now let’s create another folder - logs
This folder may sometimes be the reason your code doesn’t run. Just mkdir logs
It is also really important to import the augustus folder to the same directory.
Augustus = /shared/pkg/augustus - you could symlink this to the same directory. (‘ln -s /shared/pkg/augustus’)
** Or if you’re in another assembly folder worked on Dr. Stajich or others you can cp -r that folder to your directory.
Finally you’ll be creating your pipeline folder. It’s best if you copy this entire folder from another worked through Assembly folder. (cp -r)
## 04- Let’s Assemble!
Dr. Stajich has played with different assemblers over the years. But his main brain child is the AAFTF wrapper. 
*wrappers are code that helps put a bunch of other code together to be run sequentially.
#AAFTF- Automatic Assembly For The Fungi - https://github.com/stajichlab/AAFTF
Steps are described below:
- AAFTF trim Trim FASTQ input reads - with BBMap
- AAFTF filter Filter contaminanting reads - with BBMap
- AAFTF assemble Assemble reads - with SPAdes
- AAFTF vecscreen Vector and Contaminant Screening of assembled contigs - with BlastN based method to replicate NCBI screening
- AAFTF sourpurge Purge contigs based on sourmash results - with sourmash
- AAFTF rmdup Remove duplicate contigs - using minimap2 to find duplicates
- AAFTF pilon Polish contig sequences with Pilon - uses Pilon
- AAFTF sort Sort contigs by length and rename FASTA headers
- AAFTF assess Assess completeness of genome assembly
- AAFTF pipeline Run AAFTF pipeline all in one go.
This is one script that I have been using that is the latest iteration of it. If you did not copy the pipeline folder from another directory- copy and paste this code into a empty txt file and call it 01_AAFTF.sh
Script begins ———
#!/usr/bin/bash
#SBATCH -N 1 -n 8 --mem 64gb --time 72:00:00 --out logs/AAFTF_new.%a.log
#above configurations are important for slurm. We're telling how much power and memory to use. How long to run (72 hours) and where to save the progress report (logs).
MEM=64
CPU=$SLURM_CPUS_ON_NODE
N=${SLURM_ARRAY_TASK_ID}
#This N is where the array comes into play. You'll be starting from the first line 1-(lastline)
if [ -z $N ]; then
N=$1
if [ -z $N ]; then
echo "Need an array id or cmdline val for the job"
exit
fi
fi
module load AAFTF
FASTQ=input
SAMPLEFILE=samples.csv
ASM=genomes
WORKDIR=working_AAFTF
#Dr. Stajich likes to save things into variables to be used later. We've mentioned that our FASTQ is the input folder where our reads are
#SAMPLEFILE is the file we just created
#ASM will be directed to genomes- this is where you will find your assembled genome, WORKDIR will be your temporary file directory.
mkdir -p $ASM $WORKDIR
#we're telling the script to create these two folders
if [ -z $CPU ]; then
CPU=1
fi
# Notice this is my script and I've adjusted my sample file to have BASE, GENUS, SPECIES, ORDER, PHYLUM saved. If you use this script make sure you've accounted for this.
IFS=, # set the delimiter to be ,
sed -n ${N}p $SAMPLEFILE | while read BASE GENUS SPECIES ORDER PHYLUM
do
ASMFILE=$ASM/${BASE}.${GENUS}.${SPECIES}.spades.fasta
VECCLEAN=$ASM/${BASE}.${GENUS}.${SPECIES}.vecscreen.fasta
PURGE=$ASM/${BASE}.${GENUS}.${SPECIES}.sourpurge.fasta
CLEANDUP=$ASM/${BASE}.${GENUS}.${SPECIES}.rmdup.fasta
PILON=$ASM/${BASE}.${GENUS}.${SPECIES}.pilon.fasta
SORTED=$ASM/${BASE}.${GENUS}.${SPECIES}.sorted.fasta
STATS=$ASM/${BASE}.${GENUS}.${SPECIES}.sorted.stats.txt
LEFTIN=$FASTQ/${BASE}_1.fastq.gz
RIGHTIN=$FASTQ/${BASE}_2.fastq.gz
LEFTTRIM=$WORKDIR/${BASE}.${GENUS}.${SPECIES}_1P.fastq.gz
RIGHTTRIM=$WORKDIR/${BASE}.${GENUS}.${SPECIES}_2P.fastq.gz
LEFT=$WORKDIR/${BASE}.${GENUS}.${SPECIES}_filtered_1.fastq.gz
RIGHT=$WORKDIR/${BASE}.${GENUS}.${SPECIES}_filtered_2.fastq.gz
echo "$BASE $GENUS $SPECIES"
# We just created a bunch of variables, these will all be used below. A point for you to focus on is the LEFTIN and RIGHTIN variables.
# Sometimes your reads will be in XX_R1.fq.gz XX_R2.fq.gz
# Notice how different that is from XX_1.fastq.gz and XX_2.fastq.gz. Make sure to look at your input files and see if these match- if not nano into it and change it.
# The rest of the script don't worry about it too much. As time goes on you can make more finer tuned suggestions.
if [ ! -f $ASMFILE ]; then # can skip we already have made an assembly
if [ ! -f $LEFT ]; then # can skip filtering if this exists means already processed
if [ ! -f $LEFTTRIM ]; then
AAFTF trim --method bbduk --memory $MEM --left $LEFTIN --right $RIGHTIN -c $CPU -o $WORKDIR/${BASE}.${GENUS}.${SPECIES}
fi
AAFTF filter -c $CPU --memory $MEM -o $WORKDIR/${BASE}.${GENUS}.${SPECIES} --left $LEFTTRIM --right $RIGHTTRIM --aligner bbduk
if [ -f $LEFT ]; then
rm $LEFTTRIM $RIGHTTRIM # remove intermediate file
fi
fi
AAFTF assemble -c $CPU --left $LEFT --right $RIGHT --memory $MEM \
-o $ASMFILE -w $WORKDIR/spades_${BASE}.${GENUS}.${SPECIES}
if [ -s $ASMFILE ]; then
rm -rf $WORKDIR/spades_${BASE}.${GENUS}.${SPECIES}/K?? $WORKDIR/spades_${BASE}.${GENUS}.${SPECIES}/tmp
fi
if [ ! -f $ASMFILE ]; then
echo "SPADES must have failed, exiting"
exit
fi
fi
if [ ! -f $VECCLEAN ]; then
AAFTF vecscreen -i $ASMFILE -c $CPU -o $VECCLEAN
fi
if [ ! -f $PURGE ]; then
AAFTF sourpurge -i $VECCLEAN -o $PURGE -c $CPU --phylum $PHYLUM --left $LEFT --right $RIGHT
fi
if [ ! -f $CLEANDUP ]; then
AAFTF rmdup -i $PURGE -o $CLEANDUP -c $CPU -m 500
fi
if [ ! -f $PILON ]; then
AAFTF pilon -i $CLEANDUP -o $PILON -c $CPU --left $LEFT --right $RIGHT
fi
if [ ! -f $PILON ]; then
echo "Error running Pilon, did not create file. Exiting"
exit
fi
if [ ! -f $SORTED ]; then
# AAFTF sort -i $PILON -o $SORTED
AAFTF sort -i $VECCLEAN -o $SORTED
fi
if [ ! -f $STATS ]; then
AAFTF assess -i $SORTED -r $STATS
fi
done
…
Stop once you get to done and paste into your empty text file.
To run this script make sure you are in your main directory you are working from.
What’s does that look like now?
ls -l
Should look like:
..augustus/
..input/
..logs/
..pipeline/
..samples.csv
You should see the above files. If you don’t- Make sure you cd yourself here. Now to run do:
sbatch --array=1-(what was the total lines when you ran wc -l samples.csv?) pipeline/01_AAFTF.sh (Hit ENTER)
An actual example:
sbatch --array=1-25 pipeline/01_AAFTF.sh
Running your script through an array allows multiple iterations of different data all at the same time.
Check if your script is running ok with squeue -u $YOURUSERNAME
..
Good luck!
-T